Research
We use high performance computing to model the physical behaviour of biological macromolecules with the ultimate aim of addressing biological questions. We have the unusual philosophy that the underlying physics of biomolecular interactions is key to truely understanding their behaviour.
Research topics
- Novel Continuum Algorithms for Mesoscale Biosimulation: Fluctuating Finite Element Analysis (FFEA)
- Expansion of Fluctuating Finite Element Analysis: Applications to Cytoplasmic Dynein
- Understanding Fibrin Protofibrils at the Molecular Level
- Insights into the Molecular Mechanisms for Myosin VII regulation
- Calculating Configurational Entropies from Molecular Dynamics
- Understanding Supercoiling-Dependent DNA Recognition: A Combined Experimental and Computational Approach
- DNA Supercoiling and Topology
Novel continuum algorithms for mesoscale biosimulation: Fluctuating Finite Element Analysis (FFEA)
(Robin Oliver, Robin Richardson, Sarah Harris, Daniel Read, Oliver Harlen)
Modeling the dynamics of proteins is a very computationally expensive task. A wide variety of simulation techniques exist, a popular example being Molecular Dynamics (a method used extensively within this research group). However, such a model typically involves detailed simulation of the protein's structure at, or near to, the atomic level. It is therefore unsuitable for modelling many biological entities, in particular molecular motors composed of large protein domains (e.g. Myosin, Kinesin and Dynein), or systems in which many very large proteins are interacting (e.g. the crowded cytoplasm). Furthermore, there are many proteins whose atomistic structures are as yet unknown, and for which the only structural information available is the overall shape obtained through experimental techniques such as Cryo-EM (see the EMDataBase) or SAXS.
Our meso-scale protein model treats globular proteins as a viscoelastic continuum subject to thermal noise [1]. The protein is discretised into a tetrahedral mesh, and the dynamics due to thermal fluctuations are resolved through Finite Element Analysis; consequently this new algorithm is called Fluctuating Finite Element Analysis (FFEA) . The model is parameterised locally by the bulk, continuum properties of each region of the protein: namely the shear viscosity, bulk viscosity, shear elasticity, bulk elasticity and density. The model also accommodates intermolecular interactions such as van der Waals interactions and electrostatics, which enables the dynamics of multiple coarse-grained proteins to be modelled. The animation below shows an FFEA simulation of the molecular motor dynein, based on the 3D cryo-EM reconstruction from our collaborator Stan Burgess [2].
Our ultimate aim is to fully develop FFEA into a user-friendly software tool for protein simulation that we will make available to the biophysics community.
1. Oliver R., Read D. J., Harlen O. G. & Harris S. A. "A Stochastic Finite Element Model for the Dynamics of Globular Macromolecules" J. Comp. Phys. 239, 147-165, 2013.
2. Roberts AJ; Malkova B; Walker ML; Sakakibara H; Numata N; Kon T; Ohkura R; Edwards TA; Knight PJ; Sutoh K; Oiwa K; Burgess SA "ATP-driven remodeling of the linker domain in the dynein motor". Structure 20 1670-1680, 2012.


Expansion of Fluctuating Finite Element Analysis: Applications to Cytoplasmic Dynein
(Ben Hanson, Sarah Harris, Daniel Read, Oliver Harlen)
The function of the molecular motor cytoplasmic dynein is to walk along microtubules, carrying cargo towards the cell nucleus [1]. It achieves this via an ATP driven cycle of conformational changes and binding events. Simulating the mechanisms of these events requires knowledge of the internal structure, but also of interest are the kinetics of the molecule [2] which occur over time scales accessible to FFEA.
We are currently in the process of introducing a general kinetic scheme into FFEA to simulate these conformational changes. By creating a linear mapping between the pre and post conformational states, together with an associated rate, we are able to switch between the two structures mid simulation. This gives us the ability to see exactly how exploration of the energy landscape of the system of proteins (via conventional FFEA) affects the rates at which those proteins undergo conformational changes. The scheme is general enough that we envisage being able to simulate the shape dependent fluctuations, interactions and kinetics of any mesoscale protein.
1. Roberts AJ; Kon T; Knight PJ; Sutoh K; Burgess SA "Functions and mechanics of dynein motor proteins". Nat. Rev. Mol. Cell Biol. 14 713-726, 2013.
2. Sarlah A; Vilfan A; "The winch model can explain both coordinated and uncoordinated stepping of cytoplasmic dynein". Biophys. J. 107 662-671, 2014.

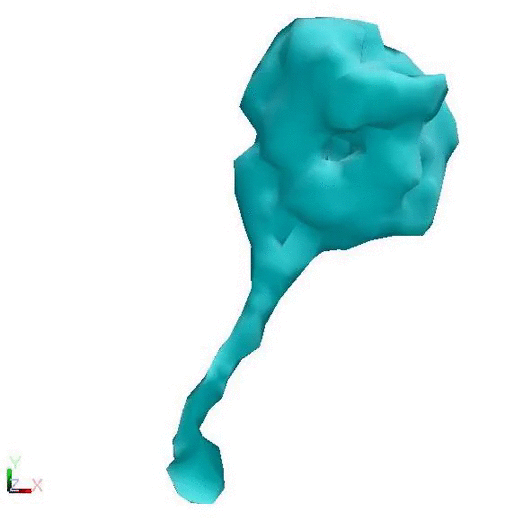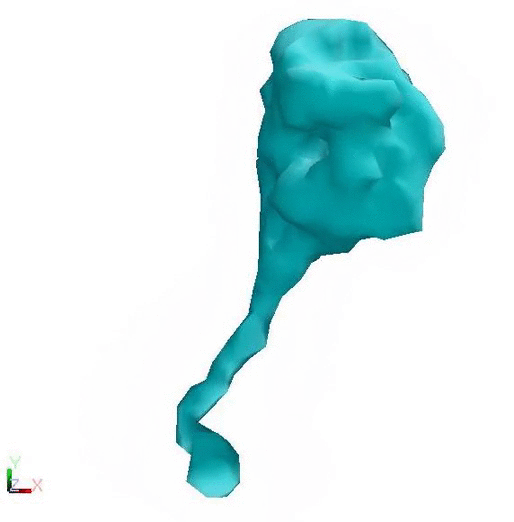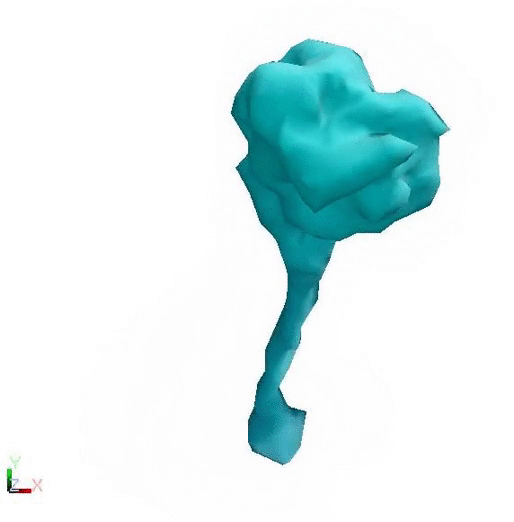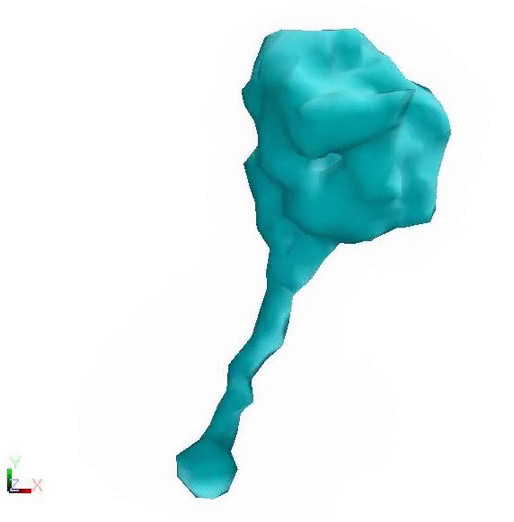
Post Powerstroke - Lower flexibility Pre Powerstroke - Higher flexibility

Cytoplasmic dynein changing conformation
Understanding Fibrin Protofibrils at the Molecular Level
(Albert Solernou)
 Understanding Fibrin Protofibrils at the Molecular Level (PDF)
Understanding Fibrin Protofibrils at the Molecular Level (PDF)
Insights into the Molecular Mechanisms for Myosin VII regulation
(Glenn Carrington)
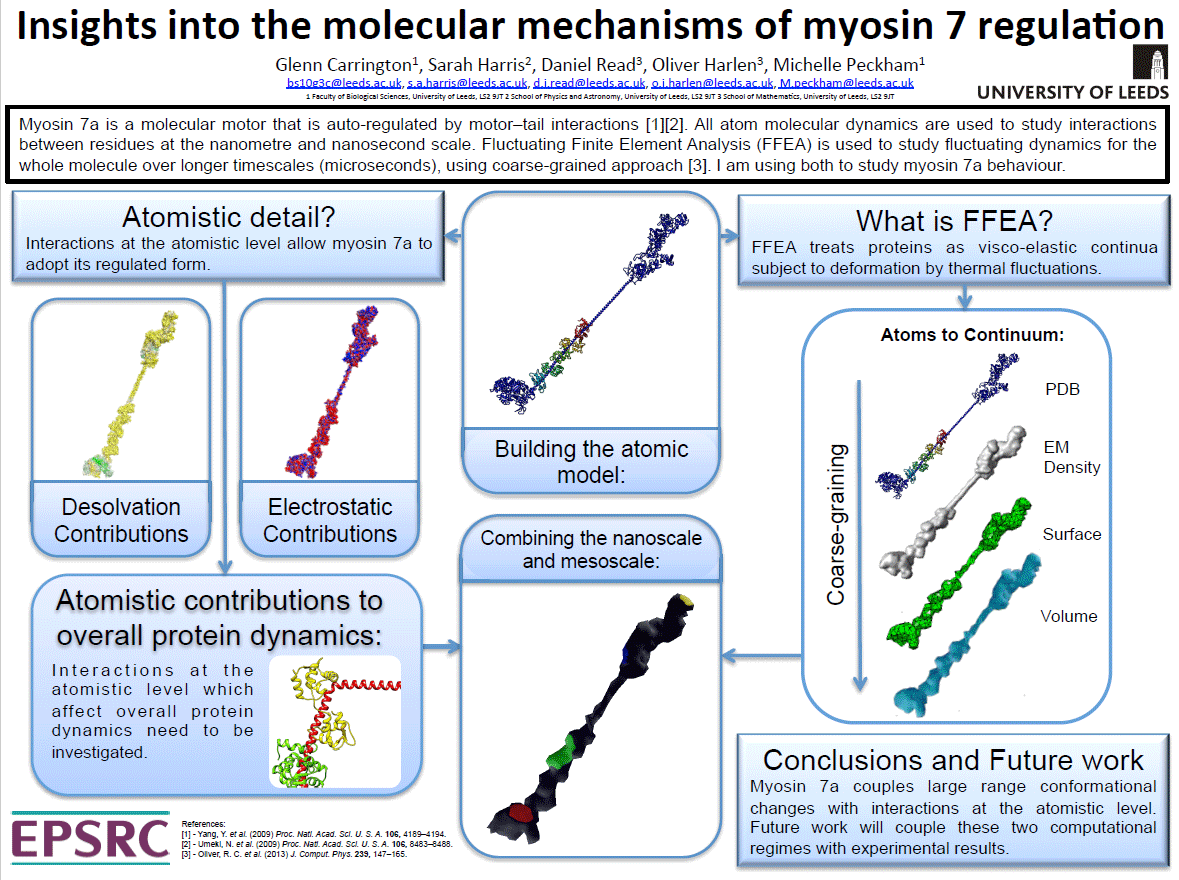 Insights into the Molecular Mechanisms for Myosin VII regulation (PDF)
Insights into the Molecular Mechanisms for Myosin VII regulation (PDF)
Calculating Configurational Entropies from Molecular Dynamics
(Outi Kamarainen)
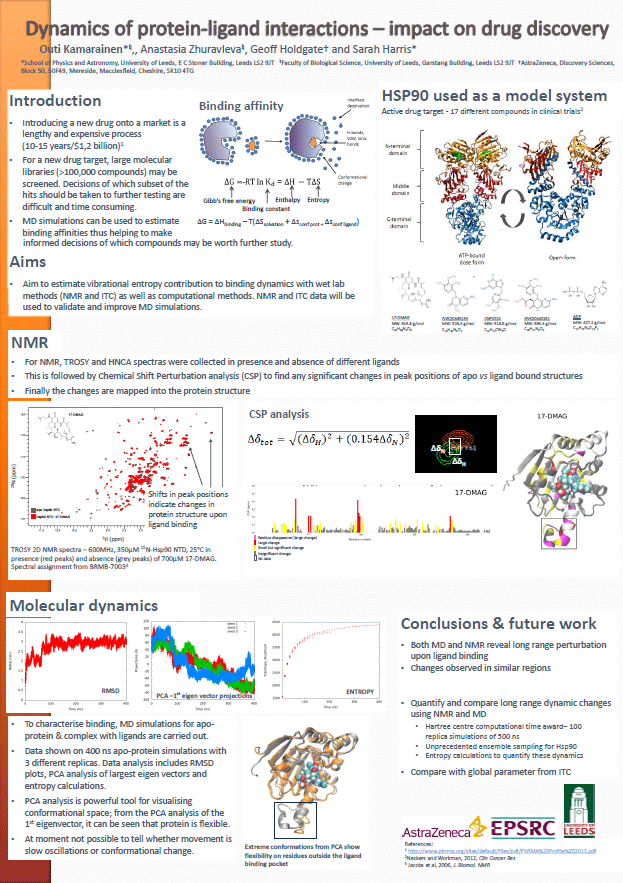 Calculating Configurational Entropies from Molecular Dynamics (PDF)
Calculating Configurational Entropies from Molecular Dynamics (PDF)
Understanding Supercoiling-Dependent DNA Recognition: A Combined Experimental and Computational Approach
(Agnes Noy and Sarah Harris)
This work is funded by the BBSRC in collaboration with Tony Maxwell at the JIC, Norwich.
Supercoiling is known to act as a global regulator of transcription; it changes the energy required to separate the two DNA strands, it affects the gross and the fine structure of the helix and thus recognition by DNA-binding proteins, while it can bring together two distant sites and hence facilitate target site location [1]. In spite of its relevance to gene regulation, the details of the structure of supercoiled DNA and how it influences DNA-protein interactions are not understood. In the current project we are studying supercoiling-dependent DNA recognition using a combination of experimental and computational approaches.
The preparation of small DNA circles of ca. 200-700 bp from E. coli LZ54 strain containing triplex binding sequences and 434 operator sites is currently underway in the Maxwell laboratory. DNA triplex formation is very sensitive to levels of supercoiling and has been developed commercially as a reporter assay for topoisomerase activity [2]. On the other hand, 434 repressor binding to DNA is strongly favoured by high twist values of DNA twist [3]. We are investigating the interactions of small DNA circles with these targets as a function of different levels of DNA supercoiling using a variety of biophysical techniques and atomistic molecular dynamics simulations. We aim to calculate the balance between twist and writhe, together with thermodynamic parameters with the final goal to provide a quantitative molecular level understanding of, and ability to control, the molecular recognition of DNA by supercoiling.
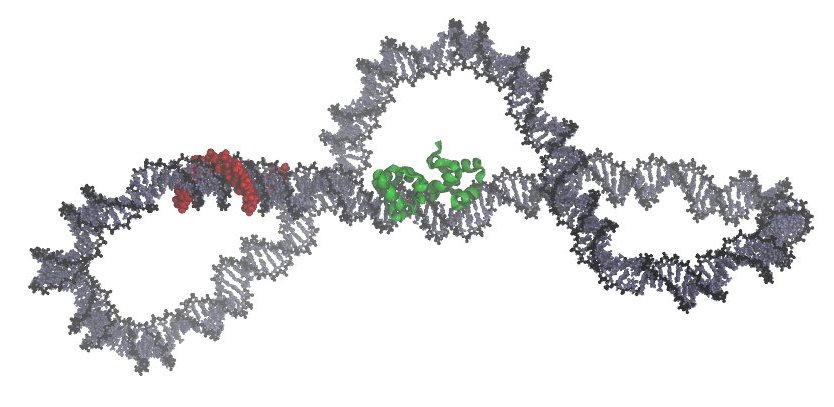
The image below shows a snapshot from our simulations of a DNA minicircle containing a bound triplex strand (in red) and the 434 repressor (in green).
1. J. M. Fogg, G. L. Randall, B. M. Pettitt, D. L. Sumners, S. A. Harris & L. Zechiedrich. "Bullied no more: when and how DNA shoves proteins around" Quart. Rev. Biophys. 102, 587-596, 2012.
2. A. Maxwell, N. P. Burton & N. O'Hagan. "High-throughput assays for DNA gyrase and other topoisomerases" Nuc. Acids Res. 34, e104, 2006.
3. G. Koudelka. "Recognition of DNA structure by 434 repressor" Nuc. Acids Res. 26, 669, 1998.
DNA Supercoiling and Topology
(Thana Sutthibutpong, Nicola Stonehouse, Sarah Harris)
DNA in vivo rarely exists as a relaxed linear molecule. In bacteria, it is generally maintained in an under-wound, supercoiled state. This supercoiling is distributed between writhe and untwisting, where the conversion between the two is crucial in allowing DNA to be both stored and transcribed. We use atomistic molecular dynamics to simulate DNA minicircles (65 - 400 bp) with different topologies to understand how the shape of the DNA depends upon the superhelical stress. The simulations show that under sufficiently high levels of superhelical stress the DNA will denature, resulting in single stranded regions [1]. We are currently investigating the sequence dependence of this denaturation for the smallest DNA loops, as shown below.

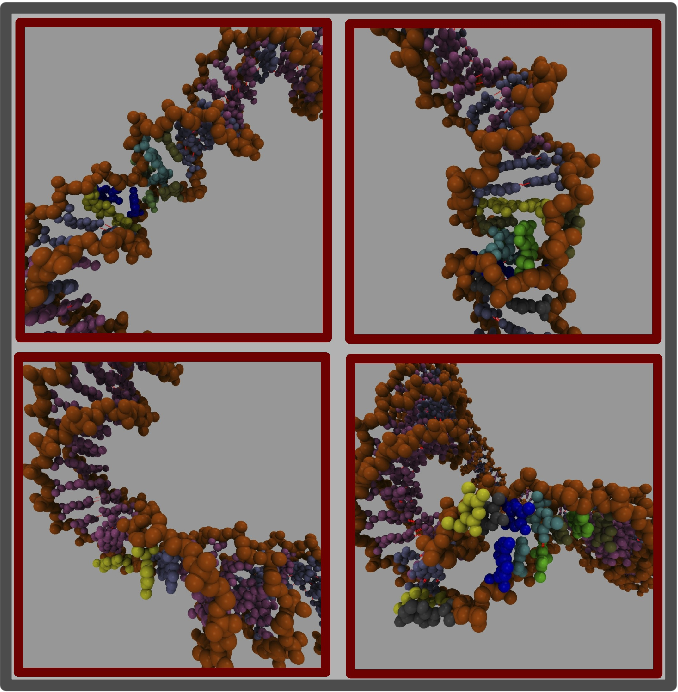
We are also using simulations of larger DNA circles (see below) to investigate the partitioning between twist and writhe, with the aim of quantifying the level of DNA compaction achieved by a given level of supercoiling in collaboration with Lynn Zechiedrich, who pioneered the experimental production of large quantities of small DNA circles [2].
1. Mitchell J. S., Laughton C. A. & Harris S. A. "Atomistic simulations reveal kinks, bubbles and wrinkles in supercoiled DNA." Nuc. Acids Res. 39, 3928-3938, 2011.
2. Jonathan M. Fogg, Natalia Kolmakova, Ian Rees, Sergei Magonov, Helen Hansma, John J. Perona and E. Lynn Zechiedrich. "Exploring writhe in supercoiled minicircle DNA." J. Phys.: Condens. Matter 18 S145-S159, 2006.

