Computational Biophysics Group
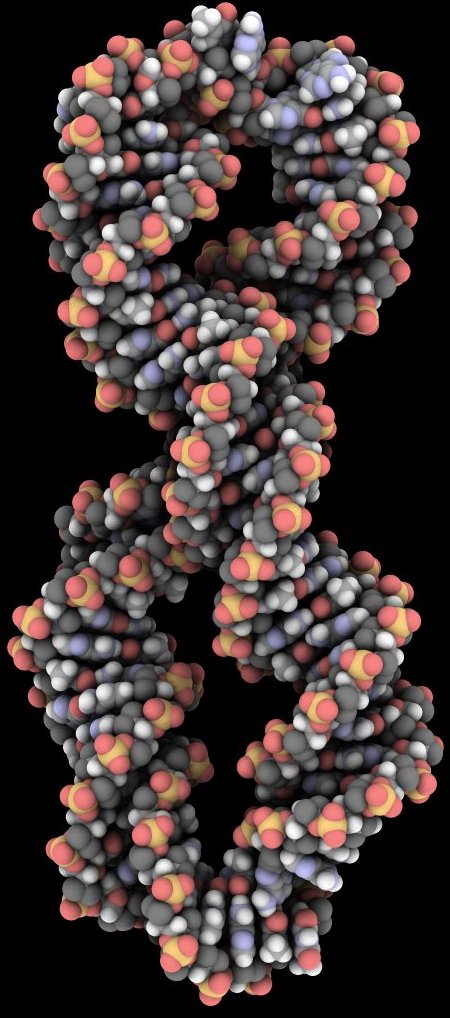
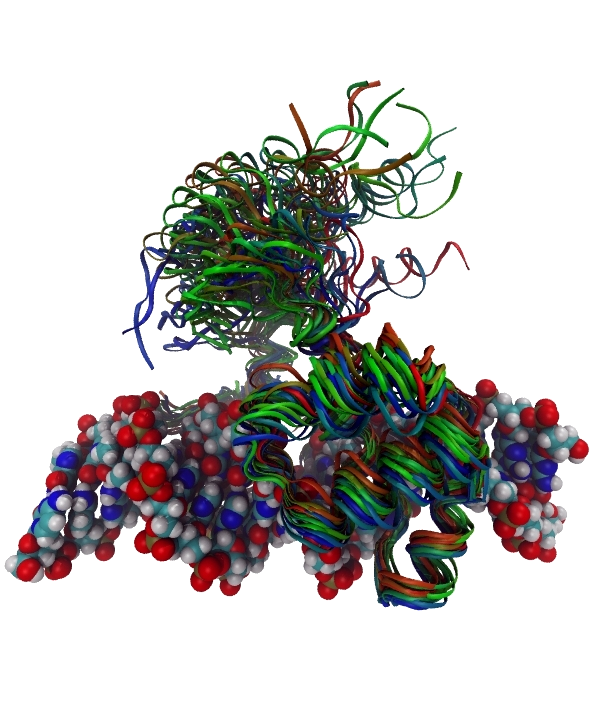
Welcome to the Computational Biophysics Group at the University of Leeds which is a part of the Soft Matter and Theoretical Physics Groups within the school of Physics & Astronomy .
We study the dynamic behaviour of proteins and nucleic acids using computer modelling and simulation. We incorporate high performance computing to model the physical behaviour of biological macromolecules with the ultimate aim of addressing biological questions. As computational models at the atomistic level allow statistical physics and thermodynamics to be combined with models that are chemically accurate, they have huge potential to provide insight into molecular biology that cannot be obtained by experiment alone.
Our current research areas include:
- Atomistic molecular dynamics (MD) simulations of DNA circles to understand the role of DNA topology and supercoiling in genetic control.
- Using a combination of atomistic MD simulation and statistical mechanics to develop new theoretical methods for quantifying the configurational entropy of biomolecules, and gaining new physical insight into how this contributes to molecular recognition, specifically for computational drug design.
- The development of a new mesoscale computational algorithm for protein modelling known as Fluctuating Finite Element Analysis to expand the range of length and time-scales that can be explored with computer simulation.